
By Malcolm Kendrick
In the last article I looked at a couple of techniques used by pharmaceutical companies to ensure that their drugs look highly beneficial. First, define the disease you are trying to treat in such a way that anything the drug does appears to be a great success. The magic trick. ‘The Prestige.’
Once done, you ruthlessly expand the market by ensuring that ever more people are diagnosed with the ‘disease’ you want doctors to treat. At this point I shall humbly quote myself from my book ‘Doctoring Data.’
“In fact, a study done in Norway a few years ago looked at the issue of cholesterol and blood pressure targets in more detail. Using guidelines developed by the European Society of Cardiology (ESC) they established that, by the age of 50, over 95% of people would have a cholesterol level, or blood pressure level, considered high enough to require drug treatment. This is despite the fact that the Norwegians are amongst the healthiest and longest-lived people on the planet. So, God knows where that leaves the rest of us.”
Yes, ninety-five percent of the population over fifty must take medications, for the rest of their lives. You’re having a laugh…no, sorry, you’re actually serious.
Another highly successful technique is to make sure that surrogate end-points can be used to claim success. What, you may ask, is a surrogate end-point?
It may be best to start by looking at clinical end-points? In a drug trial, a clinical end point will be to demonstrate something like a reduction in heart attacks, or kidney failure, or pain, or depression or… almost anything unpleasant you can think of. Fungal nail infection, male pattern baldness.
These are the type of things you’re trying to stop happening, or conditions you’re trying to improve. To that end, you set up a randomized controlled trial with two ‘arms’, placebo vs. active drug, and off you go. These trials can take many years, needing thousands of participants. They may also cost millions, even hundreds of millions of dollars.
Although it has to be said that trials don’t need to last anything like that long. If, for example, you’re trying to prove that your drug is effective for pain relief. It may only take hours, or minutes, to show this.
Even here, things are not quite that straight forward. Before setting out to prove that your drug has a beneficial effect, you first need to show that it is safe. In that it doesn’t cause kidney failure, or wreck the liver, or something equally nasty. This requires safety and toxicity studies. Animals first, then humans. But these don’t take too much time. Weeks, or months, not years.
On the other hand, if you are trying to prove your drug reduces the risk of heart failure, or strokes, or heart attacks, the trial may need to last for several years. Because you can wait a long time for a clinical event, such as a heart attack, to take place.
And, unless, or until, you have a sufficient number of clinical events, or end-points, you will not be able claim a ‘statistically significant benefit’. Two heart attacks vs. one is not going to cut it.
Which means that clinical trials in areas such as blood pressure typically last five years. At least they used to. As did the early trials on reducing cholesterol levels, and lowering blood sugar. Five years of money draining away, and no drug to sell.
In addition to the enormous cost of running a major study, there is a clock ticking away in the background. New drugs are granted patent protection which lasts for twenty years. Once those twenty years have passed, the patent runs out. At which point, generic drug manufacturers can legally make the same drug, and sell it. Without the need to run a single clinical trial.
So, in addition to the enormous cost of the trial itself, the requirement to wait five years, or more, before you can get the drug to market represents a further enormous chunk of lost income. Five years, out of twenty.
Solution?
You work hard to gain consensus from experts that, for example, a raised cholesterol level truly is a cause of cardiovascular disease. Or, to put it another way, it is a real ‘disease’ in need of treatment. Ergo, the more you lower the cholesterol level, the greater the benefit will be. The same argument is used for lowering for blood pressure, and blood sugar levels.
If you can get the regulatory authorities to agree with this, there is no need to show your drug reduces heart attacks, and strokes. You simply need to show that it lowers the cholesterol level. You are now working with, what is known as, a ‘surrogate’ end-point. [There are other forms of surrogate end points].
You work hard to gain consensus from experts that, for example, a raised cholesterol level truly is a cause of cardiovascular disease. Or, to put it another way, it is a real ‘disease’ in need of treatment.
Given this, it’s not difficult to see why pharmaceutical companies are keen, beyond keen, to have surrogate end-points accepted as the end-point for any clinical trial. There are potentially billions upon billions of dollars at stake here.
To see this process in action we can go back in time, starting with an editorial in the American Heart Association Journal, The Cholesterol Controversy Is Over. Why Did It Take So Long?
The controversy that swirled around the cholesterol-coronary heart disease problem for so many years has been, to say the least, somewhat heated.
Today the controversy is over. Discussion now centers not on whether hypercholesterolemia* is a causative factor but rather on how we should best deal with it. In 1984, the National Institutes of Health convened a Consensus Conference on Lowering Blood Cholesterol.’
The conference panel, which I was privileged to chair, “concluded unanimously that there was a cause-and-effect relation between hypercholesterolemia and coronary heart disease (CHD) risk.”
*Hypercholesterolaemia is the scientific term for raised blood cholesterol
Yes, there was a conference in 1984 which… “concluded unanimously that there was a cause-and-effect relation between hypercholesterolemia and coronary heart disease (CHD) risk.”
Perhaps the key conclusion of the conference, from a pharmaceutical company perspective, was the following:
“Individuals with high-risk blood cholesterol should be treated intensively by dietary means under the guidance of a physician, dietitian, or other health professional; if response to diet is inadequate, appropriate drugs should be added to the treatment regimen.”
They should have said. When diet doesn’t work.
This was organized shortly after Merck began safety, toxicity, and then early clinical trials of lovastatin – the first statin to be launched. A remarkable temporal coincidence. As noted in the article ‘A historical perspective on the discovery of statins.’
“The drug dramatically reduced cholesterol levels and was well tolerated. No tumors were detected. In November 1986, Merck sent the New Drug Application (NDA) to the U.S. FDA and lovastatin was given FDA approval to become the first commercial statin in September 1987.”
Merck was never required to prove there was a reduction in any clinical end-points. All they had to do was to show the drug was safe, and that it lowered cholesterol. This was sufficient for FDA approval. Indeed, the first clinical trial on lovastatin involved fifty-nine men, and lasted four weeks.
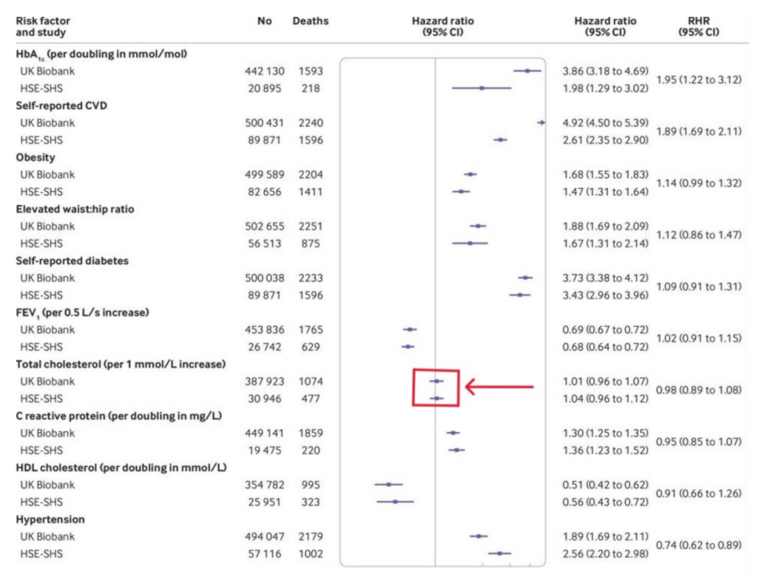
Has it ever been inarguably demonstrated, outside of drug trials, that cholesterol is a risk for cardiovascular disease? Below, I am going to show you a table from the UK Biobank study. This is possibly the largest ever study to look at risk factors for a number of different diseases, not just cardiovascular diseases. Here from their website:
“What is UK Biobank?
“UK Biobank is a large-scale biomedical database and research resource containing de-identified genetic, lifestyle and health information and biological samples from half a million UK participants.
“It is the most comprehensive and widely-used dataset of its kind, and is globally accessible to approved researchers who are undertaking health-related research that is in the public interest, whether they are from academic, commercial, government or charitable settings.
“UK Biobank is helping to advance modern medicine and enable better understanding of the prevention, diagnosis, and treatment of a wide range of serious and life-threatening illnesses – including cancer, heart disease and stroke.”
Those running the Biobank study analyzed common risk factors for cardiovascular disease, such as: smoking, sex, physical activity, blood sugar levels etc. They produced many tables and graphs.
The table below is the one with cholesterol levels vs. the risk of dying of cardiovascular disease. It established the increased risk of CVD associated with every 1mmol/l increase in blood cholesterol. [The US and Europe use different units for measuring cholesterol. In the US the unit used is mg/dl. 1mmol/l = 38.67mg/dl].
First, I want to draw your attention to the solid line in the middle of the long rectangular box. This line represents a ‘hazard ratio’ of one. Keeping this explanation for hazard ratios as simple as possible, a hazard ratio of one (HR = 1) means an average risk/hazard of cardiovascular death.
Or, to put this another way, if you have a HR of one, then you are no more, or less, likely to die of CVD than the average person of your age – all other risk factors taken into account. If the hazard ratio (HR) is below one, you have a reduced risk of cardiovascular death. If the HR is greater than one, there is an increased risk.
The ‘confidence interval’ in this table, represents the range of values that surrounds the HR. The narrower the confidence interval, the stronger the likelihood that your finding is robust (apologies to any stray statistics reading this).
To explain. If you toss a coin twice, and get heads once, and tails once, your ‘confidence’ that this coin will come up heads or tails an equal number of times is very ‘wide’. As you toss the coin more and more times, the confidence interval narrows down. After a million coin tosses, with half a million heads and half a million tails thrown, the confidence interval – that there is an equal chance of heads vs. tails – becomes very, very narrow.
Moving on. At the top of the table, you can see a high blood sugar level (as measured by HbA1c) increases the risk of CVD a great deal. It has a hazard ratio of 3.86. [Sorry, but to make things more complex, there are two different studies used in this paper. Biobank, and the smaller, combined, HSE-SHS study(s). Nothing is ever simple in the world of medical statistics. But the two studies are pretty much in agreement, about everything. Because HSE-SHS looks at fewer people, the confidence intervals are much wider. A wider ‘bar’].
Lower down, I have highlighted the Hazard Ratio associated with each 38.67mg/dl (1mmol/l) increase in blood cholesterol levels, using a big arrow pointing at a box.
As you can see, the HR for an increase in blood cholesterol was so close to one as makes no difference. Or, to put it another way, a raised cholesterol was not a risk factor for dying of cardiovascular disease. Indeed, there is not even the slightest hint of any increased risk. (HR 0.98 CI = 0.89 – 1.08). Nice, tight confidence intervals.
Compare and contrast this with the conference in 1984 which…“concluded unanimously that there was a cause-and-effect relation between hypercholesterolemia and coronary heart disease (CHD) risk”.
[CHD is a subset of cardiovascular disease. For many, the terms are interchangeable.]What did this Biobank paper have to say about their finding of absolutely no association between cholesterol and CVD?
- This is the first study to directly compare risk factor associations in UK Biobank with nationally representative cohort studies with conventional response rates.
- Associations of a wide range of risk factors with mortality outcomes showed close agreement between studies.
- Risk factor associations in UK Biobank seem to be generalisable.
The answer to the question I posed above is… nothing.
Yes, you can find studies where this is a (weak) association between CVD and cholesterol. You can also find the opposite, and any and all findings in between. Here I will quote myself again, from the paper ‘LDL-C does not cause cardiovascular disease: a comprehensive review of the current literature.’ [LDL-C = low density lipoprotein (cholesterol) the fraction of ‘cholesterol’ in the blood believed to be ‘causal’ a.k.a. ‘bad’ cholesterol]
“The idea that high cholesterol levels in the blood are the main cause of CVD is impossible because people with low levels become just as atherosclerotic as people with high levels and their risk of suffering from CVD is the same or higher. The cholesterol hypothesis has been kept alive for decades by reviewers who have used misleading statistics, excluded the results from unsuccessful trials and ignored numerous contradictory observations.”
I was a co-author of this paper.
What have the ‘experts’ said more recently about using cholesterol LDL-C as a surrogate end-point. Any change? Here from 2017: ‘Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel.’
They say that there is…:
“….a remarkably consistent dose-dependent log-linear association between the absolute magnitude of exposure of the vasculature to LDL-C and the risk of ASCVD*; and this effect appears to increase with increasing duration of exposure to LDL-C. Both the naturally randomized genetic studies and the randomized intervention trials consistently demonstrate that any mechanism of lowering plasma LDL particle concentration should reduce the risk of ASCVD events proportional to the absolute reduction in LDL-C and the cumulative duration of exposure to lower LDL-C, provided that the achieved reduction in LDL-C is concordant with the reduction in LDL particle number and that there are no competing deleterious off-target effects.”
*ASCVD = Atherosclerotic cardiovascular disease = CVD
If anything, they have doubled down in the last few years.
At this point I think it is useful to draw your attention to the conflict-of-interest statement that accompanied this paper. A conflict of interest means that an expert has worked with, and been paid money by, a pharmaceutical company, or companies. In this case, in the therapy area of cholesterol lowering. I don’t expect you to read it all.
“Conflict of interest: J.B. has received research grants from Amgen, AstraZeneca, NovoNordisk, Pfizer and Regeneron/Sanofi and honoraria for consultancy and lectures from Amgen, AstraZeneca, Eli Lilly, Merck, Novo-Nordisk, Pfizer, and Regeneron/Sanofi. E.B. has received honoraria from AstraZeneca, Amgen, Genfit, MSD, Sanofi-Regeneron, Unilever, Danone, Aegerion, Chiesi, Rottapharm, Lilly and research grants from Amgen, Danone and Aegerion. A.L.C. has received research grants to his institution from Amgen, Astra-Zeneca, Merck, Regeneron/Sanofi, and Sigma Tau, and honoraria for advisory boards, consultancy or speaker bureau from Abbot, Aegerion, Amgen, AstraZeneca, Eli Lilly, Genzyme, Merck/MSD,Mylan, Pfizer, Rottapharm and Sanofi-Regeneron. M.J.C. has received research grants from MSD, Kowa, Pfizer, and Randox and honoraria for consultancy/speaker activities from Amgen, Kowa, Merck, Sanofi, Servier, Unilever, and Regeneron. S.F. has the following disclosures for the last 12 months: Compensated consultant and advisory activities with Merck, Kowa, Sanofi, Amgen, Amarin, and Aegerion. B.A.F. has received research grants from Merck, Amgen and Esperion Therapeutics and received honoraria for lectures, consulting and/or advisory board membership from Merck, Amgen, Esperion, Ionis, and the American College of Cardiology. I.G. has received speaker fees from MSD and Pfizer relating to cardiovascular risk estimation and lipid guidelines, and consultancy/speaker fee from Amgen. H.N.G. has received research grants from Merck, Sanofi-Regeneron, and Amgen. He consults for Merck, Sanofi, Regeneron, Lilly, Kowa, Resverlogix, Boehringer Ingelheim. R.A.H. has received research grants from Aegerion, Amgen, The Medicines Company, Pfizer, and Sanofi. He consults for Amgen, Aegerion, Boston Heart Diagnostics, Gemphire, Lilly, and Sanofi. J.D.H reports honoraria/research grants from Aegerion, Alnylam, Catabasis, Lilly, Merck, Pfizer, Novartis, Regeneron, Sanofi. R.M.K is a Member, Merck Global Atherosclerosis Advisory Board. U.L. has received honoraria for lectures and/or consulting from Amgen, Medicines Company, Astra Zeneca, MSD, Berlin Chemie, Bayer, Abbott, and Sanofi. U.L. aufs has received honoraria for board membership, consultancy, and lectures from Amgen, MSD, Sanofi, and Servier. L.M. has received honoraria for consultancy and lectures from Amgen, Danone, Kowa, Merck, and Sanofi-Regeneron. S.J.N. has received research support from Amgen, AstraZeneca, Anthera, Cerenis, Novartis, Eli Lilly, Esperion, Resverlogix, Sanofi-Regeneron, InfraReDx. and LipoScience and is a consultant for Amgen, AstraZeneca, Boehringer Ingelheim, CSL Behring, Eli Lilly, Merck, Takeda, Pfizer, Roche, Sanofi-Regeneron, Kowa. and Novartis. B.G.N. reports consultancies and honoraria for lectures from AstraZeneca, Sanofi, Regeneron, Aegerion, Fresenius, B Braun, Kaneka, Amgen. C.J.P. has received research support from Roche, MSD and honoraria from MSD, Sanofi/Regeneron, Amgen and Pfizer. F.J.R. has received grants/research support from Amgen and Sanofi and has received speaker fees or honoraria for consultation from AstraZeneca, Merck, Amgen, and Sanofi. K.K.R. has received research grants from Amgen, Sanofi-Regeneron and Pfizer and honoraria for lectures, advisory boards or as a steering committee member from Aegerion, Amgen, Sanofi-Regeneron, Pfizer, AstraZeneca, Cerenis, ISIS Pharma, Medco, Resverlogix, Kowa, Novartis, Cipla, Lilly, Algorithm, Takeda, Boehringer Ingelheim, MSD. Esperion, and AbbieVie. H.S. has received research grants from AstraZeneca, MSD, Bayer Vital, sanofi-aventis, and Pfizer and honoraria for speaker fees from AstraZeneca, MSD, Genzyme, sanofi-aventis, and Synlab. He has consulted for MSD and AstraZeneca. M.R.T. has received speaker fees from Amgen, Astra Zeneca, Chiesi Pharma and Eli Lilly and speaker fees and research support from Amgen, Sanofi Aventis and Novo Nordisk. She has consulted for AstraZeneca. L.T. has received research funding and/or honoraria for advisory boards, consultancy or speaker bureau from Abbott Mylan, Actelion, Aegerion, Amgen, AstraZeneca, Bayer, Boehringer Ingelheim, Daiichi-Sankyo, GlaxoSmithKline, Menarini, Merck, Novartis, Pfizer, Sanofi-Regeneron, Servier and Synageva. G.F.W. has received research support from Amgen and Sanofi-Regeneron. O.W. has received honoraria for lectures or consultancy from Sanofi, Amgen, MSD, and Astra-Zeneca. B.v.S, and J.K.S. report no disclosures.”
Summary
Using surrogate end-points for clinical trials represents a massive boost for pharmaceutical company profits. So, they work hard, behind the scenes, to ensure that this happens. It is another key part of the ‘set-up’ to make drugs look highly beneficial. You, the public, and almost all doctors are unaware that any of this is going on.
References:
- The cholesterol controversy is over. Why did it take so long?; Daniel Steinberg, MD, PhD; Circulation, Volume 80, Issue 4, October 1989.
- Lowering Blood Cholesterol to Prevent Heart Disease; JAMA Network, April 1985.
- A historical perspective on the discovery of statins; National Library of Medicine; May 2010.
- Cholesterol-lowering effect of mevinolin, an inhibitor of 3-hydroxy-3-methylglutaryl-coenzyme a reductase, in healthy volunteers; JA Tobert et. al.; National Library of Medicine; April 1982.
- What is UK Biobank?; Website.
- Comparison of risk factor associations in UK Biobank against representative, general population based studies with conventional response rates: prospective cohort study and individual participant meta-analysis; G David Batty et. al.; The BMJ; February 2020.
- LDL-C does not cause cardiovascular disease: a comprehensive review of the current literature; Uffe Ravnskov et. al.; Expert Review of Clinical Pharmacology; October 2018.
- Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel; Brian A. Ference et. al.; National Library of Medicine; August 2017.
More From This Series
Setting Up The Trick
Research Manipulation Part I
Part 1
Deliberate Obfuscation
Research Manipulation Part III
Part 3
Falsely Reporting Success
Research Manipulation Part IV
Part 4
Looking Even Closer
Research Manipulation Part V
Part 5
Placebos – The (Not) Nothing Pill
Research Manipulation Part VI
Part 6
If Placebos Are Not True Placebos…Then What
Research Manipulation Part VII
Part 7
Placebos. Trial Blinding, and Biomarkers
Research Manipulation Part VIII
Part 8
The Replication Crisis, And Where Placebos Fit In
Research Manipulation Part IX
Part 9
An Existential Crisis?
Research Manipulation Part X
Part 10
Scottish doctor, author, speaker, sceptic

Support the Broken Science Initiative.
Subscribe today →
recent posts

Paradigm Shifts, Entrenched Thinking, and the Future of Medicine
A new paper proposes a single ratio, read off a fingerprick, that tells you where your metabolism is headed. It started in a cancer clinic. Its authors think it belongs in every MetFix affiliate.

Mark Sisson separates longevity fads from the habits that consistently improve health and performance


